Distributed Parallel Programming
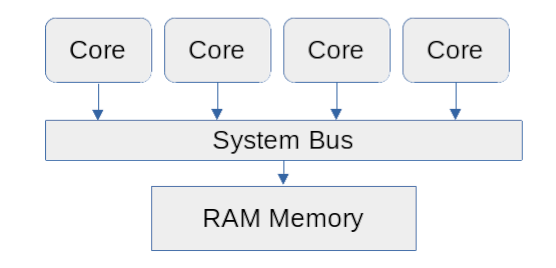
Nearly all recent computers, including personal laptops, are multicore systems. The central-processing units (CPUs) of these machines are divided into multiple processor cores. These cores share the main memory (RAM) of the computer and may share at least some of the faster memory (cache). This type of system is called a shared-memory processing or symmetric multiprocessing (SMP) computer.
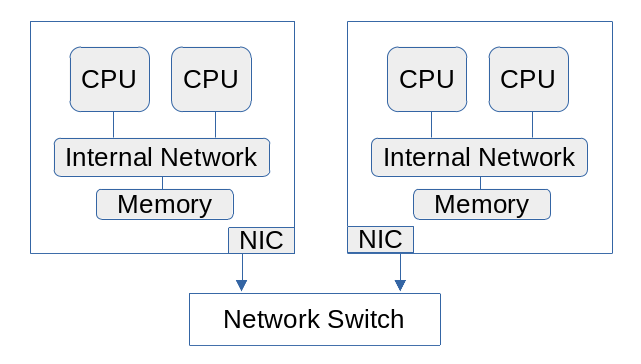
A computing cluster consists of a group of computers connected by a network. High-performance clusters nearly always have a network that is faster than the Ethernet used by consumer devices. Many use a network called InfiniBand. A cluster is thus a distributed-memory processor (DMP). Each computer, usually called a node, is independent of the others and can exchange data only through the network.
Multiprocessing works only on a single computer with multiple computing cores (SMP). If you have access to a computing cluster you can use distributed parallelization to run your program on multiple computers (DMP) as well as multiple cores per computer. This requires a communications library.
Before considering parallelizing your program, it is highly desirable to spend some time optimizing the serial version. Particularly if you can utilize NumPy and Pandas effectively, you may not need to try to parallelize the code. If you still need to do so, a well-optimized, clearly-written script will make the work much easier.
Programming Note
We have seen that Multiprocessing requires the presence of a main function or section. Other parallelization packages may or may not require this, but it never hurts to include it, and it is a good idea to structure all parallel programming scripts in this manner.
import package
def main():
parallel invocations
if __name__=="__main__":
main()
or
import package
def func1():
code
def func2():
code
if __name__=="__main__":
parallel invocations
Dask
Dask is a framework for distributed applications. It works with NumPy, Pandas, and Scikit-Learn, as well as some less common packages that have been customized to utilize it. It is primarily used to distribute large datasets.
Dask breaks the work into tasks and distributes those tasks. It can use multicore systems easily. With some care, it can also use multiple nodes on a cluster if that is available. A task may be reading a file, or doing some work on a portion of a large dataframe, or processing JSON files. To accomplish this, Dask constructs a task graph. This lays out the order in which tasks must occur. Independent tasks can be run in parallel, which can considerably speed up the time to solution. Dask is lazy and results are collected only as needed.
Our examples are taken from the Dask documentation. We cover only the basics here; a more complete tutorial can be downloaded as Jupyter notebooks.
Dask is best utilized for problems for which the dataset does not fit in memory. Some overhead is associated with it, so if your optimized NumPy or Pandas code can handle the problem, it is better to use those standard packages.
Dask Arrays
Dask arrays are much like NumPy arrays, but are decomposed into subunits that Dask generally calls chunks. In the language of parallel computing, we call this data decomposition.
>>>import dask.array as da
>>>x = da.random.random((10000, 10000), chunks=(1000, 1000))
Here we have created a two-dimensional array and broken it into 100 1000x1000 subarrays. The full-sized array is not actually created so we do not need the memory required for it. This is a general rule; avoid creating large global arrays. Large, memory-hungry arrays can be slow to process or, if they do not fit in any available memory, can make the problem impossible to solve on most computers.
Most, but not all, NumPy functions and methods are available for Dask arrays.
>>>x.mean()
This returns
dask.array<mean_agg-aggregate, shape=(), dtype=float64, chunksize=(), chunktype=numpy.ndarray>
This is because the Dask functions set up a computation but most do not execute it until told to do so. To get our answer we must invoke the compute method.
>>>x.mean().compute()
0.5000237776672359
Certain operations that require the full result, such as plotting, will automatically invoke compute but it can always be done explicitly.
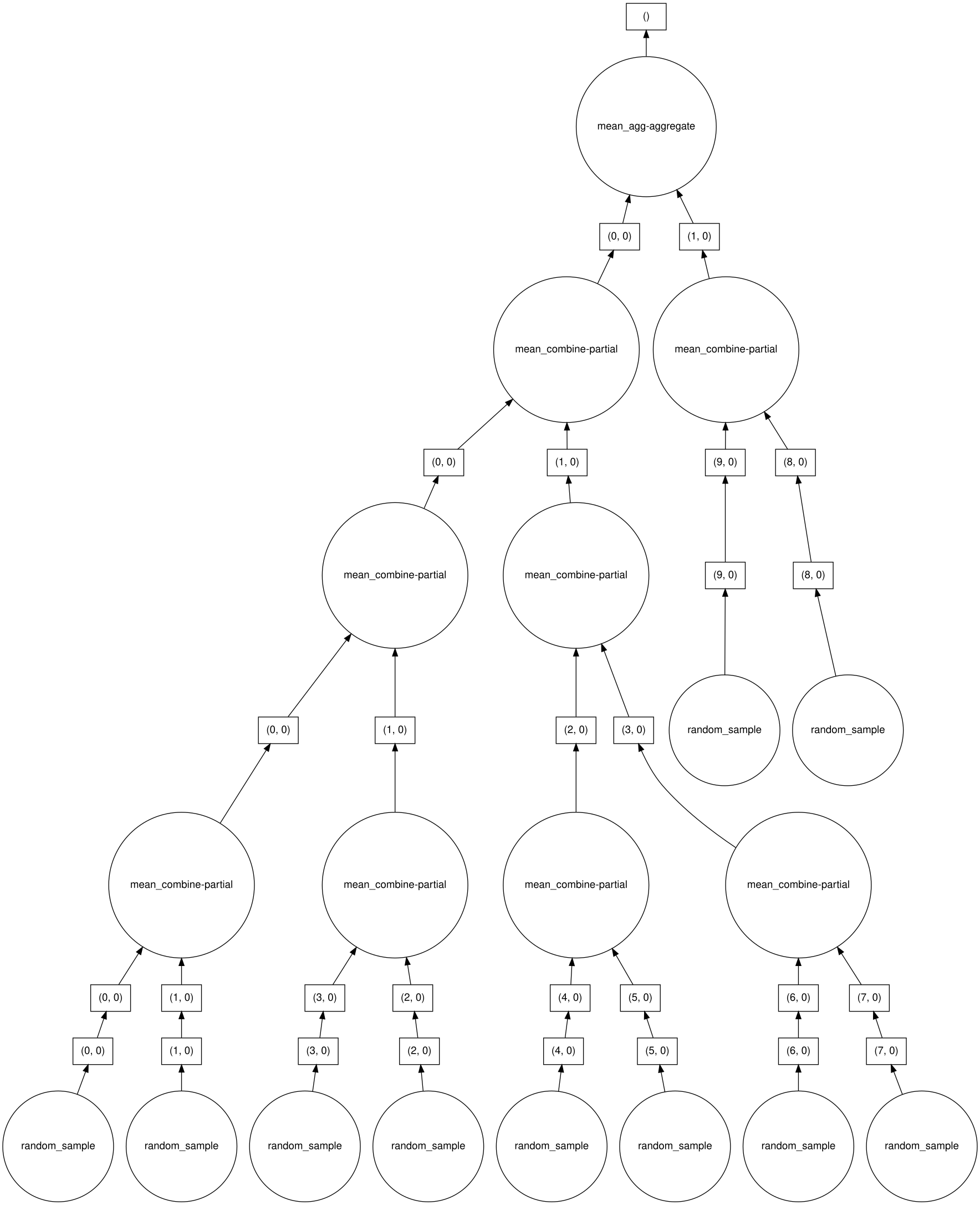
To visualize the task graph for a given Dask object, be sure that graphviz is installed (python-graphviz for conda). The visualize method for a Dask object will then produce the graph. If run at the command line, the result should be written to a file. Within a Jupyter notebook, the image will be embedded.
Example
Contents of dask_viz.py
import dask
import dask.array as da
x = da.random.random((100, 100), chunks=(10, 100))
y=x.mean()
# visualize the low level Dask graph after optimizations
y.visualize(filename="da_mean.svg", optimize_graph=True)
Expected Result
Dask Dataframes
Dask also has an infrastructure built atop Pandas. As for NumPy, many but not all Pandas functions and methods are implemented.
>>>import dask
>>>import dask.dataframe as dd
>>>df = dask.datasets.timeseries()
>>>df
This is also lazy:
Dask DataFrame Structure:
id name x y
npartitions=30
2000-01-01 int64 object float64 float64
2000-01-02 ... ... ... ...
... ... ... ... ...
2000-01-30 ... ... ... ...
2000-01-31 ... ... ... ...
Dask Name: make-timeseries, 30 tasks
Only the structure of the dataframe has been established.
We can invoke Pandas functions, but as for arrays we must finalize the results.
>>>df2 = df[df.y > 0]
>>>df3 = df2.groupby('name').x.std()
>>>df3.compute()
Exercise In a Jupyter notebook or Spyder interpreter, run the Dataframe examples above. Now run
>>>import matplotlib.pyplot as plt
or in Jupyter
%matplotlib inline
Then
>>>df[['x', 'y']].resample('1h').mean().head()
>>>df[['x', 'y']].resample('24h').mean().compute().plot()
Run
>>>plt.show()
if necessary.
The Dask version of read_csv can read multiple files using wildcards or globs.
>>>df = dd.read_csv('myfiles.*.csv')
When creating a Dataframe, Dask does attempt to assign a type to each column. If it is reading multiple files it will use the first one to make this determination, which can sometimes result in errors. We can use dtype to tell it what types to use.
>>>df = dd.read_csv('myfiles.*.csv',dtype={'Value':'float64'})
Example
Contents of dask_df_example.py
import numpy as np
import pandas as pd
import dask
import dask.dataframe as dd
df = dask.datasets.timeseries('2000','2010',freq='2H')
print(df.head())
df2 = df[df.y > 0]
df3 = df2.groupby("name").x.std()
print(df3)
print(df3.compute())
df4=df.groupby("name").aggregate({"x": "sum", "y": "max"})
print(df4.compute())
Dask Delayed
Another example of lazy evaluation by Dask is delayed evaluation. Sometimes functions are independent and could be evaluated in parallel, but the algorithm cannot be cast into an array or dataframe. We can wrap functions in delayed, which causes Dask to construct a task graph. As before, results must be requested before actual computations are carried out.
Example
Contents of dask_delayed.py
import dask
from dask.distributed import Client, progress
import time
import random
def inc(x):
time.sleep(random.random())
return x + 1
def dec(x):
time.sleep(random.random())
return x - 1
def add(x, y):
time.sleep(random.random())
return x + y
if __name__=="__main__":
client = Client(threads_per_worker=4, n_workers=1)
zs=[]
for i in range(20):
x = dask.delayed(inc(i))
y = dask.delayed(dec(x))
z = dask.delayed(add(x, y))
z=z.compute()
zs.append(z)
print(zs)
client.close()
It is also possible to invoke delayed through a decorator.
@dask.delayed
def inc(x):
time.sleep(random.random())
return x + 1
@dask.delayed
def dec(x):
time.sleep(random.random())
return x - 1
@dask.delayed
def add(x, y):
time.sleep(random.random())
return x + y
Dask Bags
Dask Bags implement collective operations like mapping, filtering, and aggregation on data that is less structured than an array or a dataframe.
Example
Download the
data. Unzip it where the Python interpreter can find it. If using Jupyter make sure to set the working folder and then provide the path to the data folder.
import dask.bag as db
b = db.read_text('data/*.json').map(json.loads)
b
b.take(2) # shows the first two entries
c=b.filter(lambda record: record['age'] > 30)
c.take(2)
b.count().compute()
Exercise
Run the above example. Why does the expression b not show any values?
Xarray
Xarray is a project that extends Pandas dataframes to more than two dimensions. It is able to use Dask invisibly to the user, with some additional keywords.
>>>import xarray as xr
>>>ds = xr.tutorial.open_dataset('air_temperature',
chunks={'lat': 25, 'lon': 25, 'time': -1})
>>>da = ds['air']
>>>da
The result shows the chunks layout but not the actual data. Without the chunks argument when the dataset was initiated, we would have an ordinary Xarray dataset.
Xarray is particularly well suited to geophysical data in the form of NetCDF files, but can also handle HDF5 and text data.
Dask and Machine Learning
Machine learning is beyond our scope here, but we will make a few comments. Dask can integrate with scikit-learn in the form of Dask-ML. You may need to use pip to install dask-ml.
>>>import numpy as np
>>>import dask.array as da
>>>from sklearn.datasets import make_classification
>>>X_train, y_train = make_classification(
n_features=2, n_redundant=0, n_informative=2,
random_state=1, n_clusters_per_class=1, n_samples=1000)
>>>X_train[:5]
>>>N = 100
>>>X_large = da.concatenate([da.from_array(X_train, chunks=X_train.shape)
for _ in range(N)])
>>>y_large = da.concatenate([da.from_array(y_train, chunks=y_train.shape)
for _ in range(N)])
>>>X_large
>>>from sklearn.linear_model import LogisticRegressionCV
>>>from dask_ml.wrappers import ParallelPostFit
>>>clf = ParallelPostFit(LogisticRegressionCV(cv=3), scoring="r2")
>>>clf.fit(X_train, y_train)
>>>y_pred = clf.predict(X_large)
>>>y_pred
>>>clf.score(X_large, y_large)
Dask can also be used with Pytorch. See the documentation for an example.