Summary and resources
Summary
- Easily develop parallel MATLAB applications without being a parallel programming expert
- Run many Simulink simulations at the same time on multiple CPU cores.
- Speed up the execution of your applications using additional hardware including GPUs, clusters and clouds
- Develop parallel applications on your desktop and easily scale to a cluster when needed
In this presentation, I’ve shown you how easy it can be to use cluster hardware with your MATLAB workflow
I’ve also shown you the specific steps needed. MathWorks provides tools and infrastructure to let you prototype on the desktop, easily start the Amazon resources you need, and extend your workflow to additional hardware.
Some Other Valuable Resources
- MATLAB Documentation
- Parallel and GPU Computing Tutorials
- Parallel Computing on the Cloud with MATLAB
MATLAB Central Community
Every month, over 2 million MATLAB & Simulink users visit MATLAB Central to get questions answered, download code and improve programming skills.
MATLAB Answers : Q&A forum; most questions get answered in only 60 minutes
File Exchange : Download code from a huge repository of free code including tens of thousands of open source community files
Cody : Sharpen programming skills while having fun
Blogs : Get the inside view from Engineers who build and support MATLAB & Simulink
ThingSpeak : Explore IoT Data
And more for you to explore…
Get Help

Quick access to:
- Self-serve tools
- Community knowledge base
- Support engineers
Part II: Matlab Parallel Computing On Rivanna
-
Upload the file Rivanna.zip to your Rivanna account using any of the methods outlined in the following link
-
Log into Rivanna using the FastX web interface and launch the Mate Desktop
-
If you are off-grounds, you will have to run the UVA Anywhere VPN before logging in through FastX
-
In the Mate Desktop, open two terminal windows, one to start the Matlab Desktop and one to work from the Rivanna command line.
-
In the second terminal window, go to the location where you uploaded the
Rivanna.zipfile and unzip it with the commandunzip Rivanna.zipto create a folder Rivanna. -
In the first terminal window, load and launch the Matlab Desktop with the commands,
module load matlab;
matlab
-
Make the current directory for Matlab the Rivanna folder and open
example1.slurmfile in the Matlab editor. -
Modify the slurm script with your own id and allocation, save it. Do this for each of the slurm example files.
-
In the second terminal window, submit the slurm script to run on Rivanna with the command, e.g.
sbatch example1.slurm -
Check that the job is in the queue with the command:
squeue -u <your id> -
Once the job is running, it should finish in a few minutes. The error file (.err) should not contain anything and the output file (.out) should contain what Matlab would normally send to the Matlab command window.
-
If for some reason you need to cancel your job, use the
scancelcommand with the job id. See further documentation here -
For the examples of running Matlab using multiple compute nodes, those jobs are submitted from within Matlab rather than with an external slurm script. Matlab creates its own slurm script to submit the job. See further documentation here
Using multiple cores on one node:
#!/bin/bash
# This slurm script file runs
# a multi-core parallel Matlab job (on one compute node)
#SBATCH -p standard
#SBATCH -A hpc_build
#SBATCH -t time=1:00:00
#SBATCH -mail-type=END
#SBATCH [--mail-user=teh1m@virginia.edu](mailto:--mail-user=teh1m@virginia.edu)
#SBATCH --job-name=runParallelTest
#SBATCH -o output=runParallelTest_%A.out
#SBATCH -e error=runParallelTest_%A.err
#SBATCH --nodes=1 # Number of nodes
#SBATCH --ntasks-per-node=8 # Number of cores per node
# Load Matlab environment
module load matlab
# Create and export variable for slurm job id
export slurm_ID="${SLURM_JOB_ID}"
# Set workers to one less that number of tasks (leave 1 for master process)
export numWorkers=$((SLURM_NTASKS-1))
# Input parameters
nLoops=400; # number of iterations to perform
nDim=400; # Dimension of matrix to create
# Run Matlab parallel program
matlab -nodisplay -r \
"setPool1; pcalc2(${nLoops}, ${nDim}, '${slurm_ID}'); exit;"
SLURM Script end-of-job email
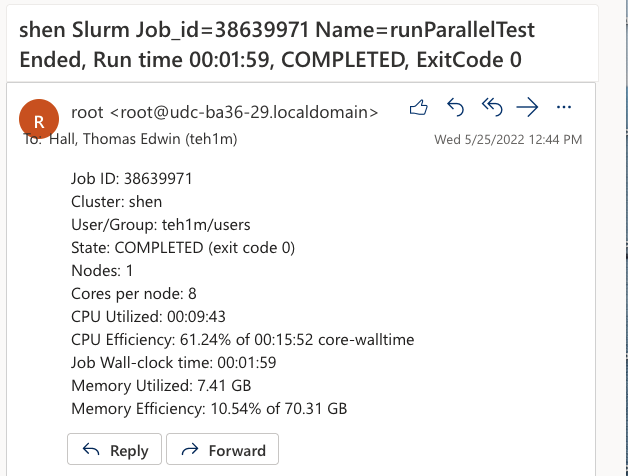
When the job finishes, the end-of-job email sent by SLURM will contain the output of the SLURM seff command.
GPU Computations
The gpu queue provides access to compute nodes equipped with RTX2080Ti, RTX3090, A6000, V100, and A100 NVIDIA GPU device
#!/bin/bash
#SBATCH -A mygroup
#SBATCH --partition=gpu
#SBATCH --gres=gpu:1
#SBATCH --ntasks=1
#SBATCH --time=12:00:00
module load singularity tensorflow/2.10.0
singularity run --nv $CONTAINERDIR/tensorflow-2.10.0.sif myAI.py
The second argument to gres can be rtx2080, rtx3090, v100, or a100 for the different GPU architectures. The third argument to gres specifies the number of devices to be requested. If unspecified, the job will run on the first available GPU node with a single GPU device regardless of architecture.
NVIDIA GPU BasePOD™
As artificial intelligence (AI) and machine learning (ML) continue to change how academic research is conducted, the NVIDIA DGX BasePOD, or BasePOD, brings new AI and ML functionality to Rivanna, UVA’s High-Performance Computing (HPC) system. The BasePOD is a cluster of high-performance GPUs that allows large deep-learning models to be created and utilized at UVA.
The NVIDIA DGX BasePOD™ on Rivanna and Afton, hereafter referred to as the POD, is comprised of:
- 10 DGX A100 nodes with
- 2TB of RAM memory per node
- 80 GB GPU memory per GPU device
- Compared to the regular GPU nodes, the POD contains advanced features such as:
- NVLink for fast multi-GPU communication* GPUDirect RDMA Peer Memory for fast multi-node multi-GPU communication
- GPUDirect Storage with 200 TB IBM ESS3200 (NVMe) SpectrumScale storage array, which makes it ideal for the following types of jobs:
- The job needs multiple GPUs on a single node or even multiple nodes.
- The job (can be single- or multi-GPU) is I/O intensive.
- The job (can be single- or multi-GPU) requires more than 40 GB GPU memory. (The non-POD nodes with the highest GPU memory are the regular A100 nodes with 40 GB GPU memory.)
Slurm script additional constraint
#SBATCH -p gpu
#SBATCH --gres=gpu:a100:X # replace X with the number of GPUs per node
#SBATCH -C gpupod
Remarks
- Before running on multiple nodes, please make sure the job can scale well to 8 GPUs on a single node.
- Multi-node jobs on the POD should request all GPUs on the nodes, i.e. –gres=gpu:a100:8.
- You may have already used the POD by simply requesting an A100 node without the constraint, since 18 out of the total 20 A100 nodes are POD nodes.
- As we expand our infrastructure, there could be changes to the Slurm directives and job resource limitations in the future. Please keep an eye out for our announcements and documentation.
Constructing Resource-Efficient SLURM scripts
- Strategy for submitting jobs to busy queues?
- Request fewer cores, less time, or less memory (and corresponding cores).
- Important to know exactly what compute/memory resources you job needs, as detailed in the seff output.
Constructing Resource-Efficient SLURM scripts
Shell script to monitor and record cpu/memory usage using top
#!/bin/bash
# This script takes four command line arguments, samples the output of the top command
# and stores the output of the sampling in the file named Top.out.
# $1 is the user ID of the owner of the processes to sample from the top output
# $2 is the name to include in the top output filename
# $3 is the top sampling interval
# $4 is the name of the code to be tracked (as shown in top)
# Example of line to include in slurm script submission before executable invoked
# ./sampleTop.sh <user_id> <filename> 10 <code_name> &
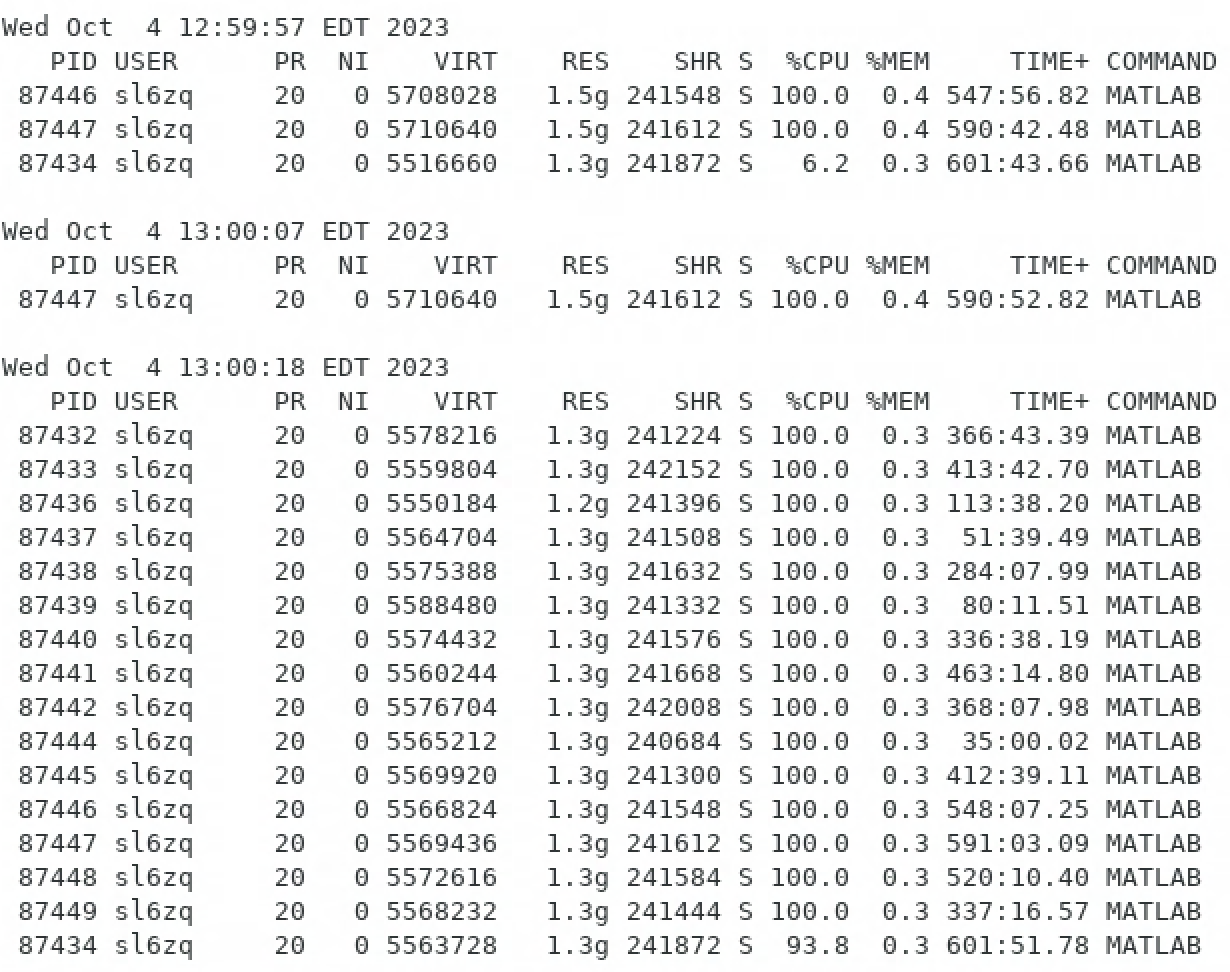
Constructing Resource-Efficient SLURM scripts
Shell script to monitor job resource usage output file
Parallel/GPU Computing
Running parallel applications
So far, we’ve covered the basics concepts of parallel computing - hardware, threads, processes, hybrid applications, implementing parallelization (MPI and OpenMP), Amdahl’s law and other factors that affect scalability.
Theory and background are great, but how do we know how many CPUs/GPUs to use when running our parallel application?
The only way to definitively answer this question is to perform a scaling study where a representative problem is run on different number of processors.
A representative problem is one with the same size (grid dimensions; number of particles, images, genomes, etc.) and complexity (e.g., level of theory, type of analysis, physics, etc.) as the research problems you want to solve.
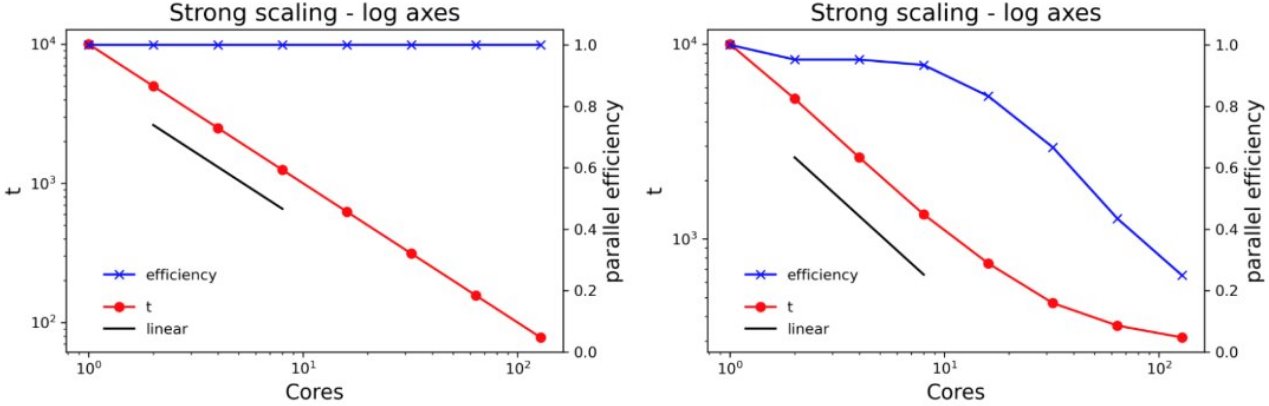
Presenting scaling results (the right way)
Plotting the same data on log axes gives a lot more insight. Note the different scales for the left axes on the two plots. Including a line showing linear scaling and plotting the parallel efficiency on the right axis adds even more value.
Where should I be on the scaling curve?
If your work is not particularly sensitive to the time to complete a single run, consider using a CPU/GPU count at or very close to 100% efficiency, even if that means running on a single core.
This specially makes sense for parameter sweep workloads where the same calculation is run many times with a different set of inputs.
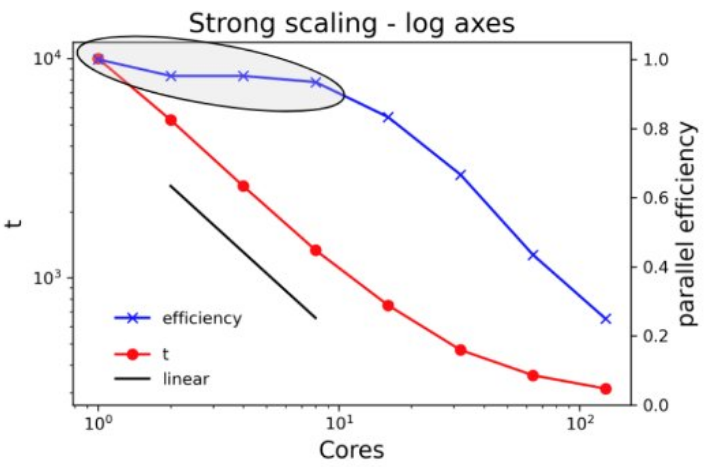
Go a little further out on the scaling curve if the job would take an unreasonably long time at lower core counts or if a shorter time to solution helps you make progress in your research.
If code does not have checkpoint-restart capabilities and the run time would exceed queue limits, you’ll have no choice but to run at higher core counts.
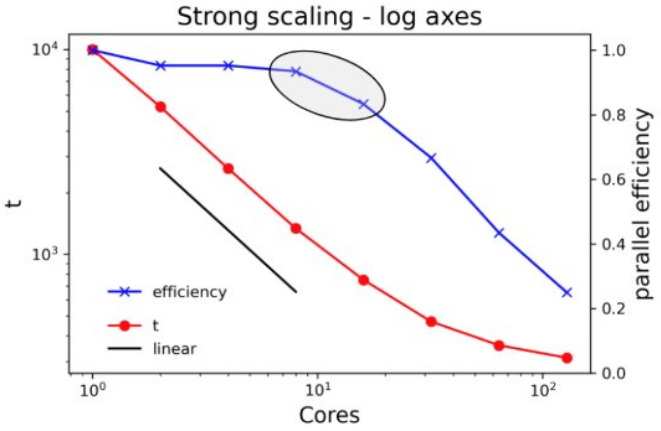
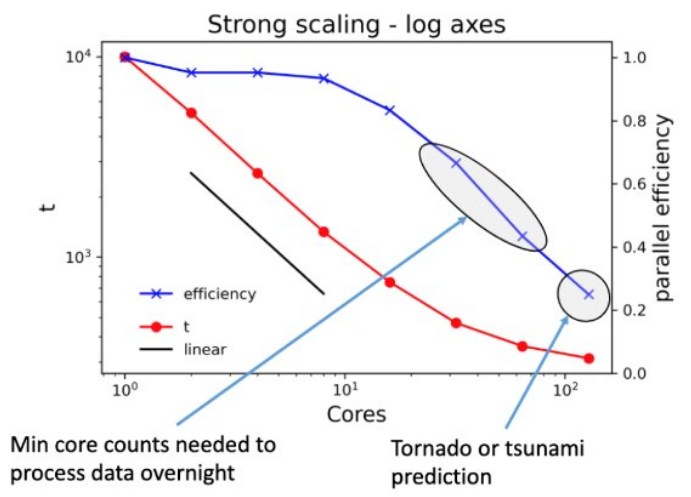
If the time to solution is absolutely critical, it’s okay to run at lower efficiency.
Examples might include calculations that need to run on a regular schedule (data collected during day must be processed overnight) or severe weather forecasting.