Batch Processing with SLURM Commands
SLURM Resource Manager

SLURM (Simple Linux Utility for Resource Management) is a resource manager (RM), also known as a queueing system. Resource managers are used to submit jobs to compute nodes from an access point generally called a frontend. Frontends are intended for editing, compiling, and very short test runs. Production jobs go to the compute nodes through the RM.
Resource Requests
A job (aka batch job) is described through a special form of shell script which contains directives to the RM to request resources. Directives are pseudocomments that take a particular form. They are read by the RM to determine the resource requirement. The job is then placed into the queue. Once a resource becomes available, the job is started on a master compute node. The master runs the job script, treating the directives as comments.
How a Scheduler Schedules Jobs
Let’s consider a simple example where there is a 6 node system, and a user wants to run 9 jobs. The scheduler places the jobs in the queue, and then on to the available nodes as they open up. Many parameters affect scheduling: number of jobs submitted, required runtime, required number of cores, required main memory, accelerators, libraries, etc.
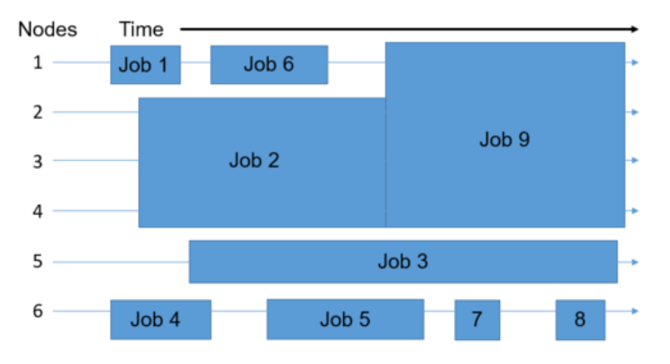
In general, every scheduler has three main goals:
- Minimize the time between the job submission and finishing the job: no job should stay in the queue for extensive periods of time
- Optimize CPU utilization: the CPUs of the supercomputer are one of the core resources for a big application; therefore, there should only be a few time slots where a CPU is not working
- Maximize job throughput: manage as many jobs per time unit as possible
For more info on how schedulers work, visit https://hpc-wiki.info/hpc/Scheduling_Basics
Batch Processing with SLURM Commands
Note that most Slurm options have two forms: a short (single-letter) form that is preceded by a single hyphen and followed by a space, and a longer form preceded by a double hyphen and followed by an equals sign. In a job script, these options are preceded by a pseudocomment #SBATCH. They may also be used as command-line options on their own.
| Option | Usage |
|---|---|
| Number of nodes | -N <n> or --nodes=<n> |
| Number of tasks | -N <n> or --ntasks=<n> |
| Number of processes (tasks) per node | --ntasks-per-node=<n> |
| Total memory per node in megabytes | --mem-per-cpu=<M> |
| Memory per node in megabytes | --mem-per-cpu=<M> |
| Wallclock time | -t d-hh:mm:ss or --time=d-hh:mm:ss |
| Partition (queue) requested | -p <part> or --partition <part> |
| Account to be charged | -A <account> or --account=<allocation> |
Note: the --mem and --mem-per-cpu options are mutually exclusive. Job scripts should specify one or the other, but not both.
Common Slurm Command Examples
Submit jobs using the sbatch command:
$ sbatch mycode-slurm.sb
Submitted batch job 8718049
Check job status using the squeue command:
$ squeue -u $USER
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
8718049 compute mycode user PD 0:00 (Priority)
Once the job is running, monitor its state with squeue:
$ squeue -u $USER
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
8718049 debug mycode user R 0:02 expanse-14-01
Cancel a running job with the scancel command:
$ scancel 8718049
SLURM Environment Variables
These are internal environment variables that exist when a job is submitted.
Input Environment Variables
Upon startup, sbatch will read and handle the options set in the following environment variables:
- SBATCH_JOB_NAME (same as
-J,--job-name) - SBATCH_ACCOUNT (same as
-A,--account) - SBATCH_TIMELIMIT (same as
-t,--time) - More…
Output Environment Variables
The Slurm controller will set the following variables in the environment of the batch script:
- SLURM_EXPORT_ENV (same as
--export) - SLURM_JOB_ID (the ID of the job allocation)
- SLURM_JOB_NAME (name of the job)
- More…
Batch Scripts Contents

Queues/Partitions
SLURM refers to queues as partitions. We do not have a default partition; each job must request one explicitly.
| Queue Name | Purpose | Job Time Limit | Memory/Node | Cores/Node |
|---|---|---|---|---|
| standard | For jobs on a single compute node | 7 days | 384 GB | 40 |
| gpu | For jobs that can use general purpose GPU’s (P100, V100, A100) | 3 days | 256 GB 384 GB 1 TB |
28 40 128 |
| parallel | For large parallel jobs on up to 50 nodes (<= 1500 CPU cores) | 3 days | 384 GB | 40 |
| largemem | For memory intensive jobs (<= 16 cores/node) | 4 days | 768 GB 1 TB |
16 |
| dev | To run jobs that are quick tests of code | 1 hour | 128 GB | 4 |
Job Arrays
A large number of jobs can be submitted through one request if all the files used follow a strict pattern. For example, if input files are named input_1.dat, …, input_1000.dat, we could write a job script requesting the appropriate resources for a single one of these jobs with:
#!/bin/bash
#SBATCH --ntasks=1
#SBATCH --time=1:00:00
#SBATCH --output-result_%a.out
#SBATCH --partition=standard
/myprogram < input_${SLURM_ARRAY_TASK_ID}.dat
In the output file name, %a is the placeholder for the array ID. We submit with:
$ sbatch --array=1-1000 myjob.sh
Many similar jobs can be submitted through job arrays. It must be a batch job.
Submit with --array=<range> option:
sbatch --array=0-30 myjobs.sh
An increment can be provided:
sbatch --array=1-7:2 myjobs.sh
# This will number them 1, 3, 5, 7
Or provide a list:
sbatch --array=1,3,4,5,7,9 myjobs.sh
Each job will be provided an environment variable SLURM_ARRAY_JOB_ID. Each task will be assigned SLURM_ARRAY_TASK_ID based on the numbers in the specified range or list.
File Specifications
It would be prudent to separate stdout and stderror:
#SBATCH -o myjobs%A%a.out
#SBATCH -e myjobs%A%a.err
Prepare files with appropriate names, e.g.:
myinput.0.in, myinput.1.in, ..., myinput.30.in
Invoke your program with a line such as:
./myexec myinput.${SLURM_ARRAY_TASK_ID}.in
Example job array script:
#SBATCH -N 1
#SBATCH -n 1
#SBATCH -t 04:00:00
#SBATCH -J montecarlo
#SBATCH -A rivanna-training
#SBATCH -p standard
#SBATCH -o output%A.%a
#SBATCH -e error%A.%a
./mccode < input${SLURM_ARRAY_TASK_ID}.dat
Using Files with Job Arrays
For more complex commands, you can prepare a file containing the text you wish to use. Your job script can read the file line by line. In the following example, you must number your subtasks starting from 1 sequentially. You must prepare the options_file.txt in advance and each line must be the options you wish to pass to your program.
#!/bin/bash
#
#SBATCH --ntasks=1
#SBATCH --partition=standard
#SBATCH --time=3:00:00
#SBATCH --array=1-1000
OPTS=$(sed -n "{SLURM_ARRAY_TASK_ID}"p options.txt)
./myprogram $OPTS
The double quotes and curly brace are required.
For more info on using job arrays, visit https://www.rc.virginia.edu/userinfo/hpc/slurm/#job-arrays
Job Dependency
With the sbatch command, you can invoke options that prevent a job from starting until a previous job has finished. This constraint is especially useful when a job requires an output file from another job in order to perform its tasks.
The –dependency option allows for the specification of additional job attributes. For example, suppose that we have two jobs where job_2.slurm must run after job_1.slurm has completed. Using the corresponding Slurm command files, we can submit the jobs as follows:
sbatch job_1.slurm
Submitted batch job 18375
sbatch --dependency=afterok:18375 job_2.slurm
Notice that the –dependency has its own condition, in this case afterok. We want job_2 to start only after the job with id 18375 has completed successfully. The afterok condition specifies that dependency. Other commonly-used conditions include the following:
after: the dependent job is started after the specified job_id starts runningafterany: the dependent job is started after the specified job_id terminates either successfully or with a failureafternotok: the dependent job is started only if the specified job_id terminates with a failure

For more info on specifying job dependencies, visit https://www.rc.virginia.edu/userinfo/hpc/slurm/#specifying-job-dependencies
More Example Slurm Scripts
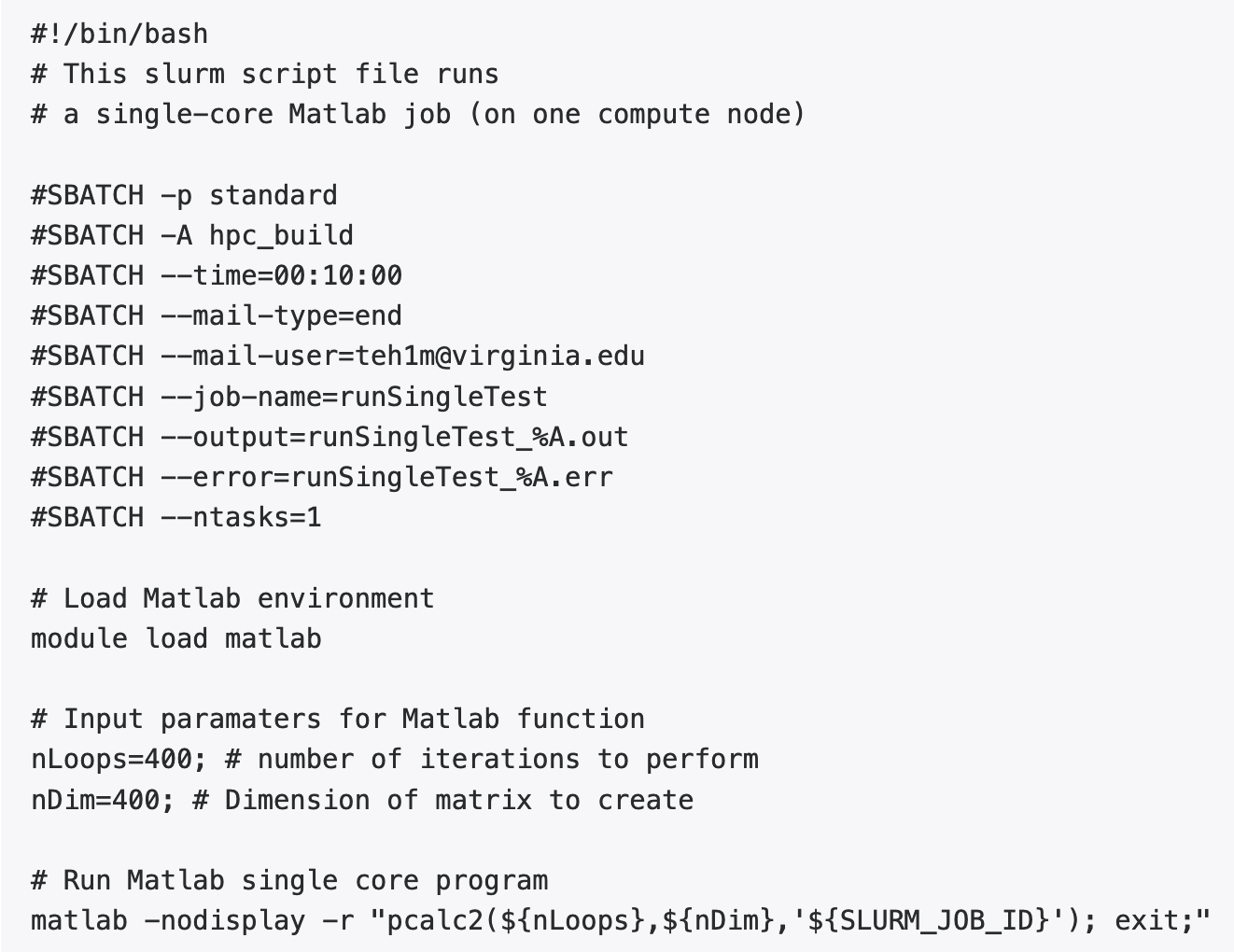
Using one core on one node:
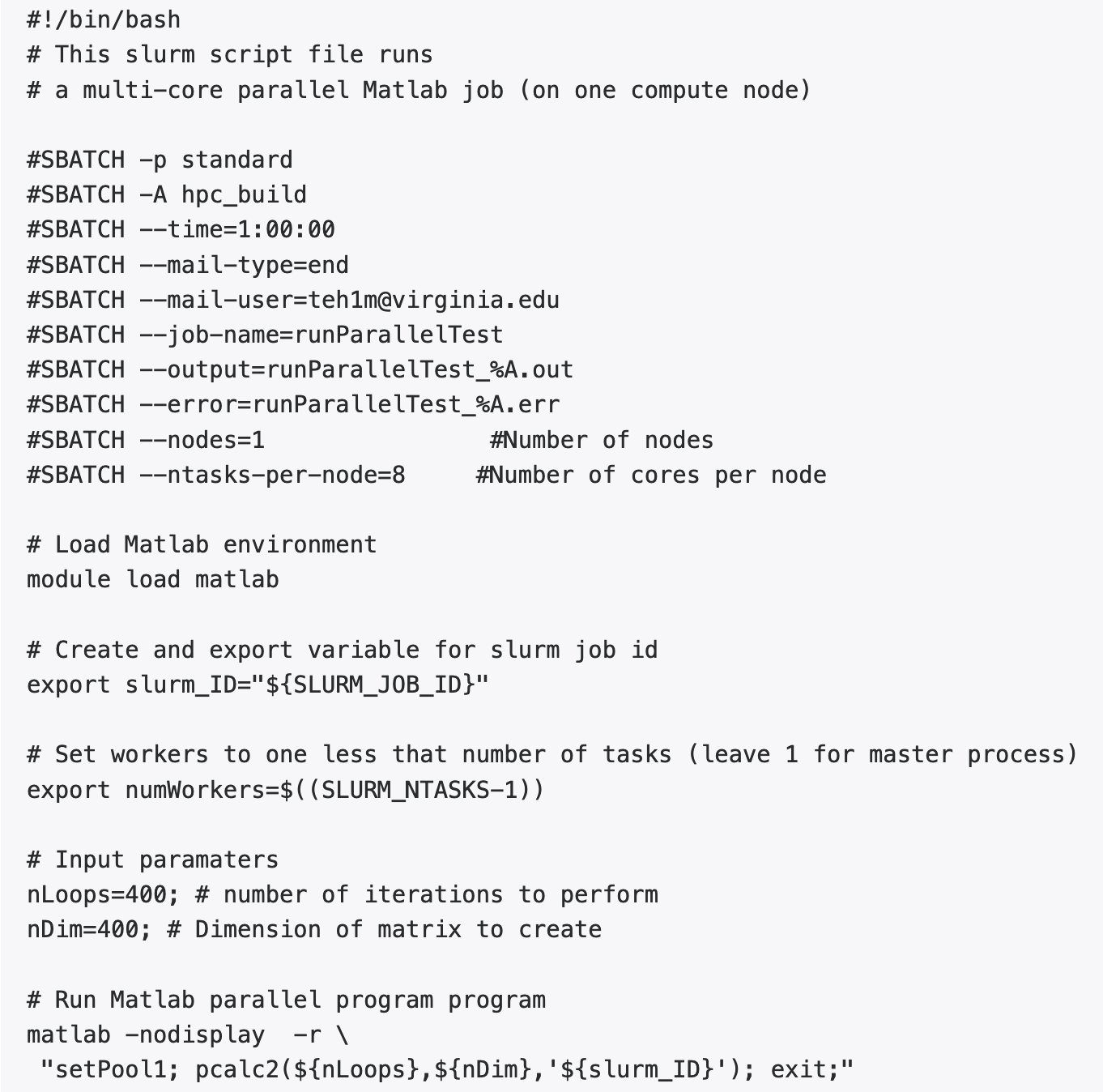
Using multiple cores on one node:
SLURM Script End-of-Job Email
When the job finishes, the end-of-job email sent by SLURM will contain the output of the SLURM seff command.

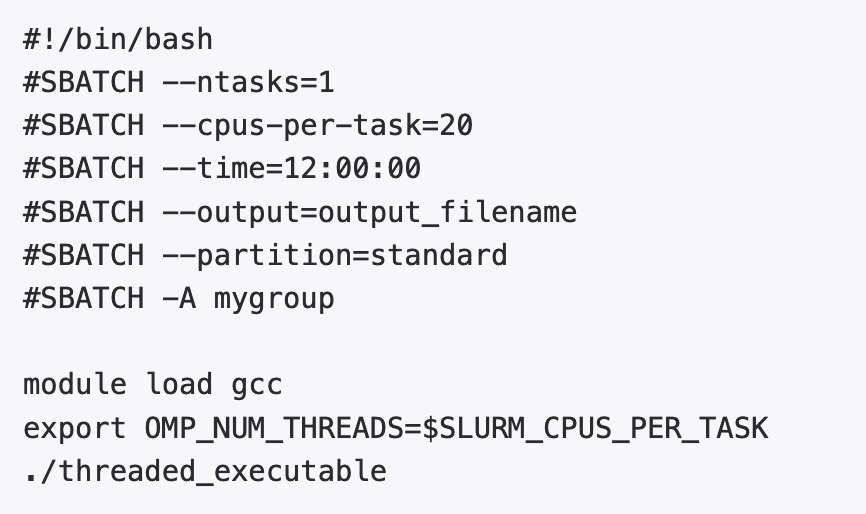
Threaded Jobs (OpenMP or Pthreads)
Slurm considers a task to correspond to a process. Specifying a number of CPUs (cores) per node ensures that they are on the same node. Slurm does not set standard environment variables such as OMP_NUM_THREADS or NTHREADS, so the script must transfer that information explicitly. This example is for OpenMP:
Distributed Memory Jobs
If the executable is a parallel program using the Message Passing Interface (MPI), then it will require multiple processors of the cluster to run. This information is specified in the Slurm nodes resource requirement. The script srun is used to invoke the parallel executable. This example is a Slurm job command file to run a parallel (MPI) job using the OpenMPI implementation:

Both OpenMPI and IntelMPI are able to obtain the number of processes and the host list from Slurm, so these are not specified. In general, MPI jobs should use all of a node, so we’d recommend ntasks-per-node=40 on the parallel partition.
GPU Computations
The gpu queue provides access to compute nodes equipped with RTX2080Ti, RTX3090, A6000, V100, and A100 NVIDIA GPU device.
The second argument to gres can be rtx2080, rtx3090, v100, or a100 for the different GPU architectures. The third argument to gres specifies the number of devices to be requested. If unspecified, the job will run on the first available GPU node with a single GPU device regardless of architecture.
NVIDIA GPU BasePOD
As artificial intelligence (AI) and machine learning (ML) continue to change how academic research is conducted, the NVIDIA DGX BasePOD, or BasePOD, brings new AI and ML functionality to Rivanna, UVA’s High-Performance Computing (HPC) system. The BasePOD is a cluster of high-performance GPUs that allows large deep-learning models to be created and utilized at UVA.
The NVIDIA GPU BasePOD on Rivanna, hereafter referred to as the POD, is comprised of:
- 10 DGX A100 nodes
- 8 A100 GPU devices and 2 TB local node memory (per node)
- 80 GB GPU memory (per GPU device)
Which makes it ideal for the following types of jobs:
- The job needs multiple GPUs on a single node or even multiple nodes
- The job (can be single or multi-GPU) is I/O intensive
- The job (can be single or multi-GU) requires more than 40 GB GPU memory (the non-POD nodes with the highest GPU memory are the regular A100 nodes with 40 GB GPU memory)
Slurm script additional constraint:

Remarks:
- Before running on multiple nodes, please make sure the job can scale well to 8 GPUs on a single node.
- Multi-node jobs on the POD should request all GPUs on the nodes, i.e.
--gres=pgu:a100:8 - You may have already used the POD by simply requesting an A100 node without the constraint, since 10 out of the total 12 A100 nodes are POD nodes.
- As we expand our infrastructure, there could be changes to the Slurm directives and job resource limitations in the future. Please keep an eye out for our announcements and documentation.
For more info on POD, visit https://www.rc.virginia.edu/userinfo/hpc/basepod/